神経セロイドリポフスチン症
神経セロイドリポフスチン症(NCL)の概要と、多くみられる病型である2(CLN2)の症状や診断についてご案内いたします。

神経セロイドリポフスチン症の概要
神経セロイドリポフスチン症(NCL)は、進行の速い小児神経変性疾患です。13,14
神経セロイドリポフスチン症(neuronal ceroid lipofuscinoses ; NCL)は、「バッテン病」とも呼ばれ、小児期や青年期における認知障害の主たる原因となっています。13,15
NCLは14のタイプに分類され、以下のような病因および臨床的特徴を有します。13
神経セロイドリポフスチン症(NCL)の分類および特徴14-17
世界的に最も多くみられる病型は神経セロイドリポフスチン症2型(CLN2)と神経セロイドリポフスチン症3型(CLN3)です。14
- 疾患*
- 発症時期・病型
- 原因遺伝子
- 関連タンパク質
- CLN1
- 古典的乳児型、遅発乳児型、若年型、成人型
- CLN1(PPT1)
- Palmitoyl protein thioesterase 1
- CLN2
-
古典的:遅発乳児型、
非古典的:乳児型、若年型、緩徐進行型SCAR7† - CLN2(TPP1)
- トリペプチジルペプチダーゼ 1
- CLN3
- 古典的若年型
- CLN3
- 膜貫通型タンパク質
- CLN4
- 成人型(常染色体優性)
- CLN4(DNAJC5)
- 可溶性システインストリングタンパク質
- CLN5
- 遅発乳児変異型、若年型、成人型
- CLN5
- 可溶性リソソームタンパク質
- CLN6
- 遅発乳児変異型、成人型(Type A Kufs病)
- CLN6
- 膜貫通型タンパク質
- CLN7
- 遅発乳児変異型
- CLN7(MFSD8)
- 膜貫通型タンパク質
- CLN8
- 遅発乳児変異型
- CLN8
- 膜貫通型タンパク質
- CLN9
- 若年変異型
- ―
- ―
- CLN10
- 古典的先天型、遅発乳児型、成人型
- CLN10(CTSD)
- カテプシンD
- CLN11
- 成人型
- CLN11(GRN)
- プログラニュリン
- CLN12
- 若年型
- CLN12(ATP13A2)
- ATP分解酵素
- CLN13
- 成人型(Type B Kufs病)
- CLN13(CTSF)
- カテプシンF
- CLN14
- 乳児型、進行性ミオクロニー発作
- CLN14(KCTD7)
- カリウムチャネルタンパク質
神経セロイドリポフスチン症2型(CLN2)は、希な進行性の小児神経変性疾患
神経セロイドリポフスチン症2型(CLN2)は、常染色体劣性遺伝形式のライソゾーム蓄積症(LSD)で、CLNのなかでも最も多く認められるタイプであり、その発症頻度は200,000新生児に1人といわれています。14,18
古典的遅発乳児型は、その表現型は異なるものの、ヨーロッパで最も多く認められます。16
古典的なものは、生後から発症までの精神運動発達は正常であることが多く、一般的には2~4歳でてんかん発作が生じます。13,19
てんかん発作を機に医療機関を受診するのが一般的ですが、多くの患者さんでは言語発達の遅延が最初の症状として表れます。20,21
6歳に達する頃には、認知機能の低下、運動失調、失明に至ります。その後は、若年期に死亡に至るまで、およそ半生において介助を必要とします。
他のNCLとは異なり、CLN2では失明に至るのは後期です。13,14,22
非古典的な病型では、発症年齢、疾患進行の速さ、症状悪化の速さなどが異なります。20,22
神経セロイドリポフスチン症2型(CLN2)の症状1
CLN2の症状は年齢を重ねるとともに複雑化、悪化の一途をたどります。また、患者自身のみならず、家族の生活の質(QOL)に与える影響は甚大です。
最終的に、CLN2患者は認知機能の低下、運動失調、失明に至り、若年期に死亡に至ります。13,14
急激な進行を伴うため、特に本剤が導入されCLN2に対する治療選択肢が広がった現在においては、早期診断がより重要です。
神経セロイドリポフスチン症2型(CLN2)の進行
疾患経過とみられる症状13,14,16,19,22,23
-
1~3歳
言語発達の遅延
-
2~4歳
非誘発発作の発症
熱性痙攣が生じることもある -
3~4歳
運動失調、
認知障害の進行、運動機能低下 -
4~5歳
難治性てんかん、ミオクローヌス、ジストニア、視力低下
-
5~6歳
車椅子生活
/寝たきり -
7~8歳
失明
-
8~12歳
死亡

3歳
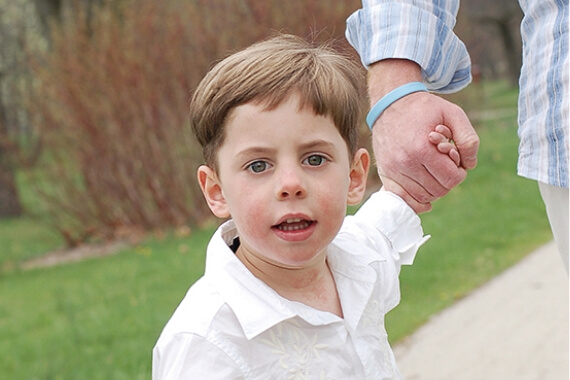
5歳
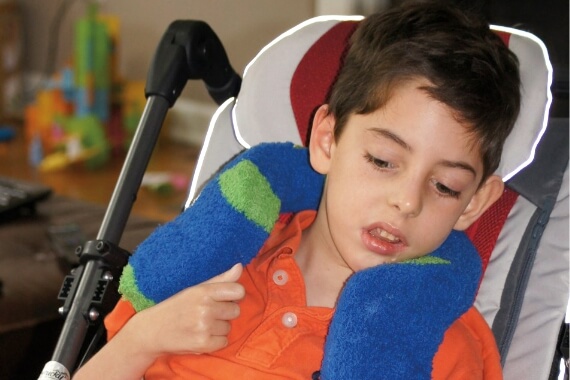
8歳
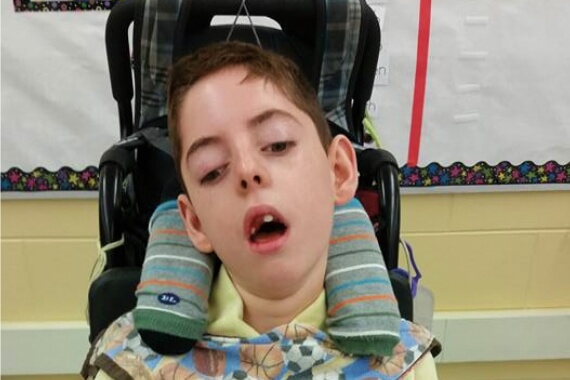
11歳
医療機関を受診する主な原因に
てんかん発作があります。
古典的な病型では2〜4歳でてんかん発作が発症しますが、それより年齢を重ねてから発症する患者も存在します。1
これらの多くは非誘発性のものですが、初回発作が熱性痙攣であることもあります。19
主にミオクロニー発作がみられますが、全般性強直間代や欠神発作、ミオクローヌス、脱力発作、間代発作、強直発作がみられることもあります。19,20,23
初期の徴候および症状
多くのCLN2患者さんでは発症初期に
言語発達遅延がみられます。21
NCL患者を登録した最大規模の多国籍症例レジストリである、DEM-CHILD registryに登録された、CLN2患者さんの疾患の特徴を定量的に評価した結果、CLN2患者の83%に言語発達遅延が認められました。
てんかん発作が認められる小児患者の診察では、CLN2に特徴的な言語発達についての詳細情報を得る必要があります。
言語発達遅延とてんかん発作はそれぞれがCLN2で多く認められ、てんかん発作が認められる前に言語発達遅延が生じていると、 CLN2が疑われます。
2~4歳の小児でこのような症状が認められる場合には、CLN2の検査を行うべきです。20,24
また、検査時の言語発達が正常の範囲内であったとしても、より若齢時の言語発達について保護者に問い合わせることが重要です。
CLN2患者に言語発達遅延が必ずしも認められるわけではありません。
初期症状としては、運動機能の低下や運動失調が現れる場合もあります。
たとえば、CLN2患者の40%では、運動機能の障害(運動失調性歩行、不器用/トリッピング、転倒しやすいなど)が認められます。21,23
自然経過
CLN2は、運動機能と言語能力の低下、急激な進行を伴う疾患です。21
DEM-CHILD registryに登録されたCLN2の古典的遅発乳児型を有する患者さんで、10年以上の観察された58例の自然経過を以下の図に示します。21,26
CLN2臨床評価尺度を用いて評価した結果、これらの患者さんにおいて運動機能と言語能力の急激な低下が認められました。21
無治療の場合、CLN2臨床評価尺度は1年間で平均2点の低下がみられました。21
運動機能と言語能力の低下21
NCL患者を登録した最大規模の多国籍症例レジストリであるDEM-CHILD registryから 収集された10年以上の自然経過21,26

神経セロイドリポフスチン症2型(CLN2)の診断
確定診断には、
酵素・遺伝子検査が必要です24
臨床検査により、CLN2の確定診断が可能となります。
最も重要な検査として、TPP1酵素活性検査と、TPP1遺伝子検査があります。24